

La pancreatitis aguda continúa siendo preocupante debido a su curso clínico incierto, pero también a que no se dispone de una terapéutica específica. El estrés del retículo endoplásmico está involucrado en etapas tempranas de su desarrollo. La modulación de las vías celulares que se activan para revertirlo podría constituir la clave en la búsqueda de futuros blancos terapéuticos, no solo para esta patología sino además para otras.
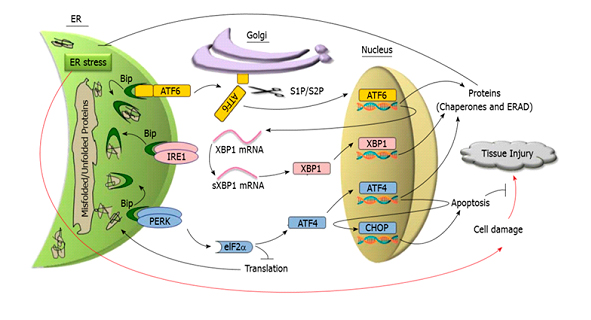
La pancreatitis aguda es una patología causada por la inflamación súbita del páncreas exocrino que resulta en la autodigestión del órgano por acción de las enzimas que este mismo produce. Se presenta con un cuadro de abdomen agudo que requiere de hospitalización inmediata. En gran parte son benignas y autolimitadas, sin embargo, en el 20% de los casos se desarrolla una forma grave que suele asociarse a falla multiorgánica con una elevada tasa de mortalidad. El curso clínico puede fluctuar imprevistamente de manera muy rápida, y no existen al presente marcadores ideales que puedan predecirlo.La pancreatitis aguda continúa siendo una gran preocupación médica no solo debido a su curso clínico incierto, sino también a que no existe una terapéutica específica para tratarla. La litiasis biliar y el abuso de alcohol son responsables en más del 80% de los casos; no obstante, se desconocen los mecanismos tempranos subyacentes al desarrollo de la patología.En la actualidad se considera que la activación in situ del tripsinógeno y del factor de transcripción nuclear kappa B (NF-κB) participan en forma independientes y complementaria en el desarrollo de la patología.El tripsinógeno, normalmente producido por el páncreas y secretado al intestino delgado, se convierte en tripsina por acción de la enzima enteroquinasa; luego, comienza el proceso necesario para degradar las proteínas en sus constituyentes fundamentales, los aminoácidos. Por su parte, el NF-κB, que fue descripto en 1986, ha concitado la atención de numerosos grupos de investigadores, en especial por sus mecanismos de regulación y su acción sobre vías inflamatorias y reguladoras de la muerte celular.En los últimos años se ha propuesto al estrés del retículo endoplásmico (RE) como un evento precedente a estos mecanismos. El RE es una organela multifuncional necesaria para la biosíntesis de lípidos, el almacenamiento de calcio y el plegado y procesamiento de proteínas estructurales o de exportación. Distintos factores, como los niveles de calcio o el estrés oxidativo, pueden perturbar la homeostasis del RE y dar como resultado una acumulación de proteínas sin plegar o bien mal plegadas en el RE, esta situación se conoce como estrés del RE.Las células acinares pancreáticas poseen la mayor tasa de síntesis proteica de todas las células del organismo por lo que son altamente susceptibles a alteraciones en la homeostasis del RE.Con el fin de restablecer la homeostasis de la organela, se dispara una respuesta a proteínas mal plegadas denominada UPR (del inglés unfolded protein response). La UPR involucra vías de señalización entre el RE y el núcleo de la célula orquestada por tres proteínas residentes del RE: PERK, IRE1 y ATF6 (figura 1). Consiste en un proceso dinámico temporal que evita, en principio, la sobrecarga del RE al detener la traducción de proteínas. Luego, se regula la expresión de genes que aumentan la capacidad de plegamiento de proteínas residentes en el RE y mejoran el control de calidad y los mecanismos de degradación de las proteínas. Sin embargo, si el estrés persiste, tanto en intensidad como en el tiempo, y estos mecanismos de supervivencia resultan insuficientes, se desencadena entonces la muerte por apoptosis para eliminar las células afectadas. La cinética de activación y la atenuación de la señal entre las proteínas sensoras de estrés pueden diferir según la naturaleza de los estímulos y el tipo celular.Estudios recientes mostraron que en la pancreatitis aguda el estrés de RE y la desregulación de la respuesta UPR juegan un papel decisivo en la patogénesis de la enfermedad en estadios muy tempranos, previos a la activación del tripsinógeno y la respuesta inflamatoria.
MECANISMOS BÁSICOS
En los últimos años se ha puesto énfasis sobre el estrés de RE y la desregulación de la UPR ya que estarían implicados en múltiples enfermedades neurológicas, metabólicas e inflamatorias, tanto en su patogénesis como en el curso de la enfermedad.
En este sentido, la vía IRE1/sXBP1 se encuentra desregulada en la esclerosis lateral amiotrófica y en la enfermedad de Huntington a través de la inhibición del proceso de autofagia. Mientras que, en la enfermedad de Alzheimer y en la enfermedad de Parkinson, XBP1 tiene un papel neuroprotector. La alteración en la vía PERK está relacionada con patologías como diabetes, enfermedades metabólicas, inflamatorias y cáncer. Finalmente, la vía ATF6 controla la gluconeogénesis en el hígado y bloquea la esteatosis inducida por estrés de RE, pero en la enfermedad de Huntington posee un papel neuroprotector.
Distintos estudios han demostrado que factores que disminuyen o revierten el estrés de RE se traducen en la atenuación de diferentes patologías.
En este sentido se demostró que el extracto de hojas de Gingko Biloba en un modelo animal de diabetes atenúa la aterosclerosis vía la inhibición de estrés de RE. En un modelo animal de ratones obesos se reportó que el ácido graso de bajo peso molecular ácido 4-fenil butírico(4-PBA) actúa como chaperona química y bloquea la adipogénesis atenuando el estrés del RE. La administración de 4-PBA posee efectos protectores en lesión isquémica cerebral mediante la inhibición del estrés de RE y la posterior vía de señalización que lleva a la apoptosis.
En un modelo animal de diabetes tipo II y en cultivo celular (Fao rat hepatoma cell) tanto 4-PBA como el ácido biliar tauroursodeoxicólico (TUDCA), que es también considerado una chaperona química, atenúan el estrés de RE y restablecen la homeostasis de la glucosa. Asimismo, se demostró que la administración de estas chaperonas reduce la activación de los principales actores de la UPR en la pancreatitis aguda experimental, atenuando significativamente la respuesta inflamatoria y la activación prematura del tripsinógeno.
La importancia de la UPR en el páncreas se refleja en los ratones knock-out para XBP1, los que muestran insuficiencia endócrina y anormalidades en la morfología del páncreas y glándulas salivales.
VÍAS DE SEÑALIZACIÓN
La UPR involucra vías de señalización entre el RE y el núcleo de la célula que es orquestada por tres proteínas transmembrana del RE: PERK (del inglés, protein kinase RNA-like endoplasmic reticulum kinase), IRE1 (del inglés inositol-requiring enzyme 1) y ATF6 (del inglés activating transcription factor 6). En la figura 1 se esquematizan los principales componentes de la UPR.
La chaperona BiP (del inglés binding immunoglobulin protein, también llamada GPR-78) es la principal proteína moduladora de la respuesta UPR. En condiciones fisiológicas BiP se encuentra asociada a las proteínas sensoras de estrés, PERK, IRE1 y ATF6, manteniéndolas inactivas. Sin embargo, bajo condiciones de estrés del RE donde proteínas sin plegar o bien mal plegadas se van acumulando progresivamente, la BiP se disocia de las proteínas transmembrana lo que induce la activación de las mismas iniciando así la UPR. Las tres proteínas transmembrana activadas estimulan distintas vías de señalización tendientes a reducir la carga de nuevas proteínas al RE, degradar proteínas mal plegadas, aumentar la capacidad de plegamiento de proteínas con el fin de restablecer la homeostasis de la organela.
La activación de PERK lleva a la fosforilación de eIF-2α (del inglés cytosolic eukaryotic translation initiation factor) que inhibe la traducción global de proteínas evitando la sobrecarga del RE. Como consecuencia de esta inhibición, ciertos ARN mensajeros que contienen pequeños marcos de lectura abierto hacia el extremo 5’ UTR (del inglés, 5′ untranslated region) son traducidos generando otras proteínas como el factor de transcripción ATF4 (del inglés activating transcription factor 4) que controla la expresión de CHOP, proteína reguladora de la transcripción de genes relacionados con la apoptosis.
La proteína sensora IRE1al activarse adquiere actividad endoribonucleasa dando origen a un potente factor de transcripción sXBP1(del inglés spliced X box-binding protein 1) que regula la síntesis de chaperonas y foldasas, induce la expansión del RE y media un proceso llamado RIDD (del inglés, IRE1 dependent decay) que atenúa la síntesis proteica. A su vez cliva micro-ARNs que controlan la expresión de la enzima caspasa-2 dando lugar a la muerte celular por apoptosis.
Al activarse, ATF6 transloca al aparato de Golgi, donde sufre una escisión proteolítica para producir un fragmento, que luego se desplaza al núcleo para promover la transcripción de los genes implicados en el ERAD (del inglés endoplasmic reticulum-associated degradation) y en el plegamiento de proteínas en el RE, como foldasas y chaperonas.
Ana Paula Courreges1, Guadalupe Álvarez21, Ana Clara Najenson3, Marcelo S. Vatta4 y Liliana G. Bianciotti5
- Instituto de Inmunología, Genética y Metabolismo (INIGEM-UBA-CONICET). ↩︎
- Instituto de Inmunología, Genética y Metabolismo (INIGEM-UBA-CONICET). ↩︎
- Instituto de Inmunología, Genética y Metabolismo (INIGEM-UBA-CONICET). ↩︎
- Instituto de Química y Metabolismo del Fármaco (IQUIMEFA-UBA-CONICET). ↩︎
- Instituto de Inmunología, Genética y Metabolismo (INIGEM-UBA-CONICET). ↩︎




